
Next-Generation Sequencing
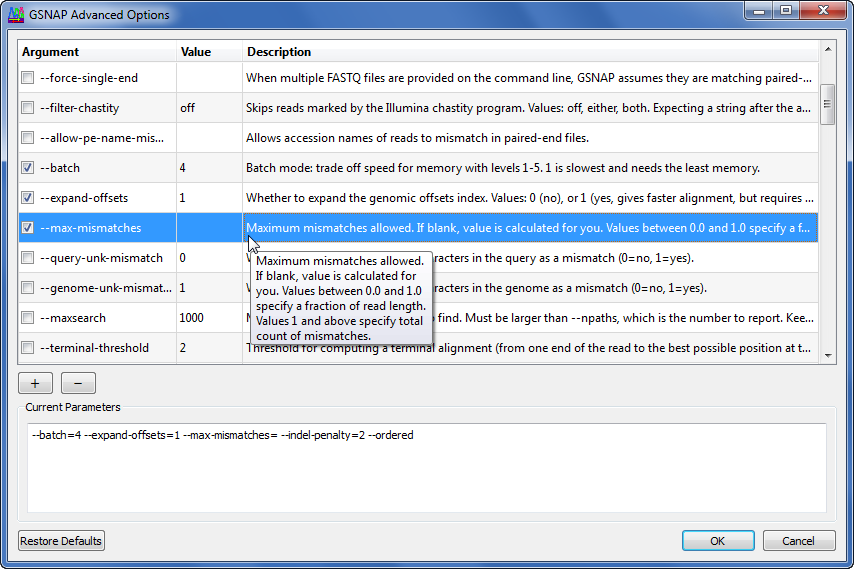
Gene Codes is committed to bringing you a wide variety of alignment and assembly algorithms developed and published in peer-reviewed journals, but making them accessible to non-engineers. For example, GSNAP (Tom Wu, Genentech, Inc.) is recognized as one of the best algorithms for correctly identifying splice junctions. A bioinformatics specialist might invoke the program like this:

Most of us work more efficiently if we have some tools to help us understand what all of the available flags and parameters are and how they affect the program. The Sequencher interface helps you select options, set values, and learn the available functionality with descriptions and tool tips. The same options from the command line above are set this way in Sequencher:
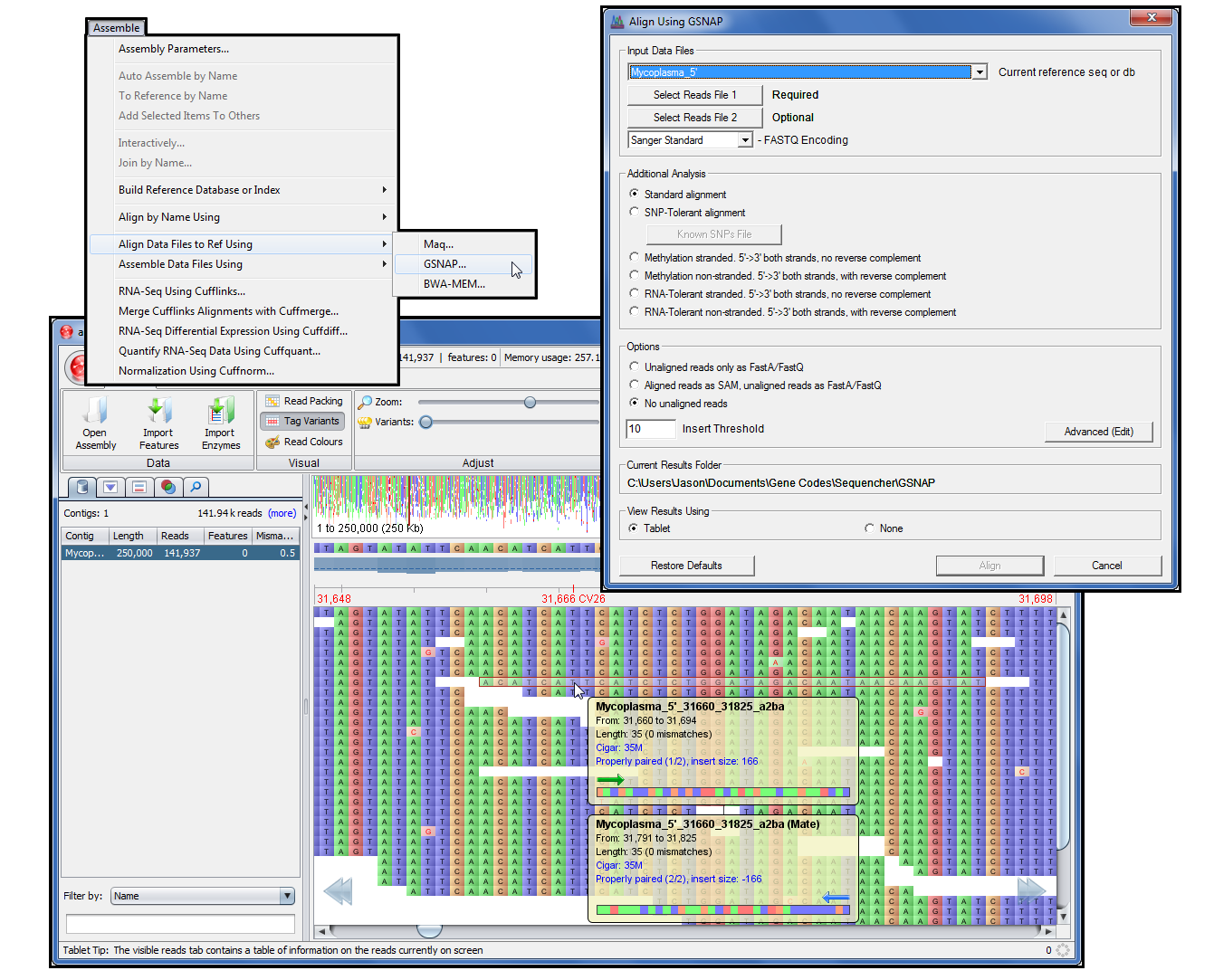
Perform SNP analysis, Methylation analysis, or RNA A-to-G tolerant alignment in just a few steps.
View your results in Tablet.
BWA, Velvet, Maq, GSNAP and Tablet are only the start for Sequencher. If you are sequencing mixed populations, then combine the power of reference-guided-alignment with de novo assembly. Align reads to a reference and capture the unaligned reads for further rounds of reference-based alignment or de novo assembly.
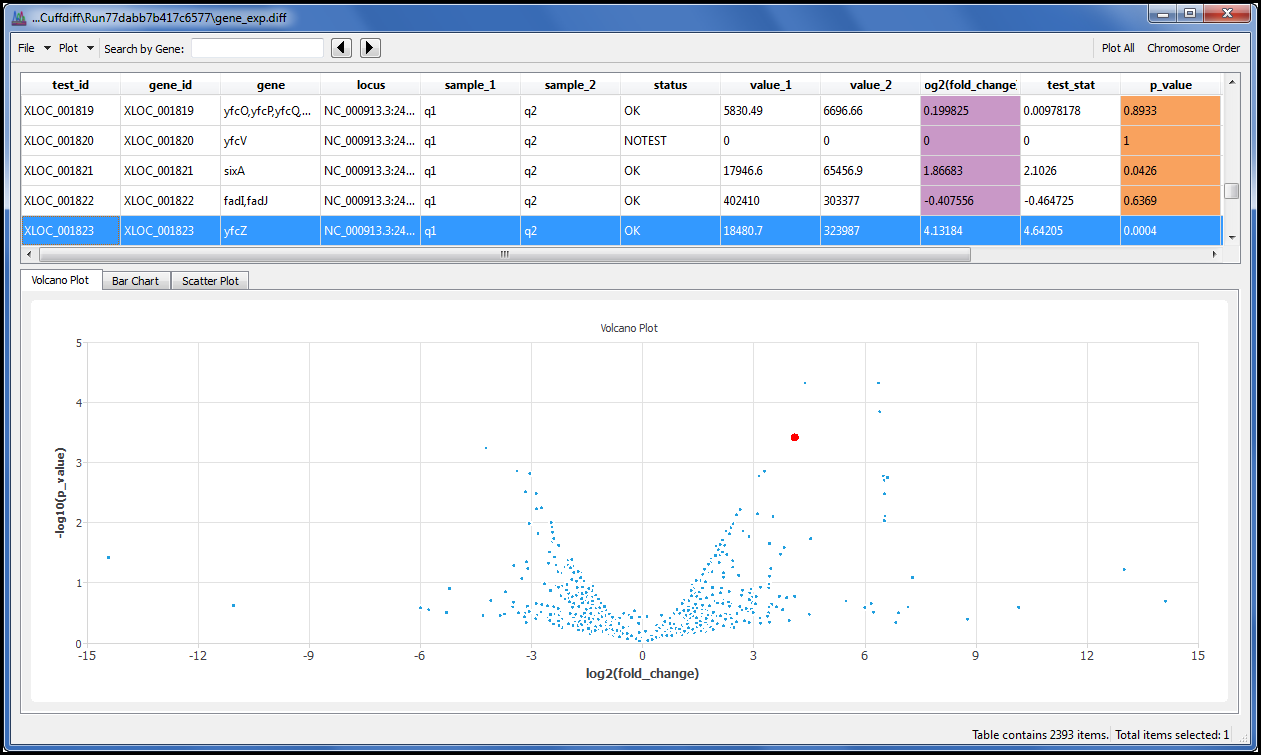
With the addition of the Cufflinks suite you can perform RNA-Seq Differential Expression using the same style of interface we developed for all our NGS algorithms.
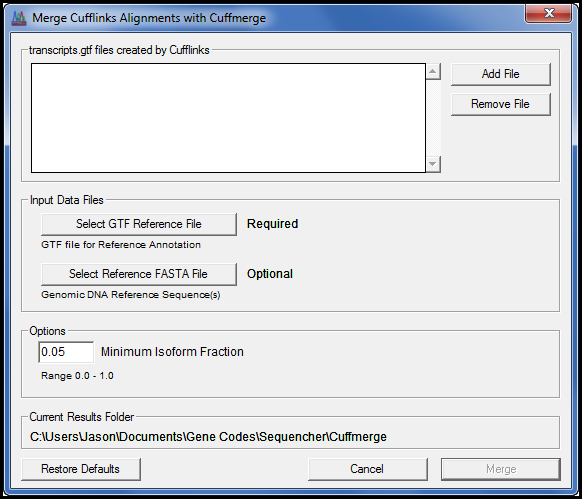
We also added the charts you would expect to see, such as the Volcano plot, so you can view the results without resorting to the command line. Because we know you have your own favourite aligners we donÔÇÖt insist you use ours. As long as your aligner creates SAM or BAM as its output you are ready to explore your data with one of the most widely regarded RNA-Seq tools available.